The equation of state relates the pressure p, volume V and temperature T of a physically homogeneous system in the state of thermodynamic equilibrium f(p, V, T) = 0. The equation called the thermic equation of state allows the expression of pressure in terms of volume and temperature p = p(V, T) and the definition of an elementary work δA = pδV at an infinitesimal change of system volume δV. As distinguished from thermic equations, the caloric equation of state specifies the dependence of the internal energy of the system E on volume V and temperature T (either p and V or p and T).
The equation of state is a fundamental characteristic of a substance which makes possible the application of the general principles of thermodynamics and hydrodynamics to particular physical objects.
Equations of state cannot be derived from thermodynamic relations alone but rely also on either experimental measurements or on theoretical calculations by the methods of statistical physics, using various models of interparticle interactions in a system. The First Law of Thermodynamics suggests that there exists a caloric equation of state while the Second, a relation between the thermodynamic and the caloric equations of state in the form ∂E/∂VT = T(∂p/∂T)V − p. The thermodynamic relationship allows the determination of the thermic and caloric equations of state, if the thermodynamic potential, specified in the form of a function of appropriate variables, is known. Thus, for instance, for Helmholtz free energy F as a function of volume V and temperature T, we have

A simple example of the equation of state for gases is the Clapeyron-Mendeleev equation pν = RT, where R is the gas constant and v is the volume of one mole. For real gases, and considering the interaction between particles, virial equations of state pν = RT(1 + B(T)/ν + C(T)/ν2 + ... ) are widely used, where B(T), C(T), ... , are the second, third, etc. virial coefficients. These coefficients are the functions of temperature alone and, depend on the forces of two, three or more particles interacting in the system. This equation of state for real gases is the most theoretically-substantiated one, but the calculation of higher order virial coefficients is limited by the difficulties presented by multiple integrals. With a rise in density, the character of the convergence of the virial expansion also deteriorates drastically. In dense systems, nonadditivity of interparticle interaction becomes significant which, when considered, changes drastically the values of the higher order virial coefficients.
A qualitatively accurate picture of a phase diagram of real gases yields the van der Waals equation of state (p + a/ν2)(ν − b) = RT, in which a and b are constants whose values are determined from the adequate description of experimental data. This equation considers both the existence of attractive forces between the gas molecules, which brings about a decrease in pressure, and the presence of repulsive forces, which appears when the distances between molecules are small, preventing an infinite compression of the gas. The van der Waals equation of state has made it possible for the first time to obtain a thermodynamically-consistent description of a phase gas-liquid transition, which terminates at the critical point with parameters pc, νc, and Tc —where the difference between the liquid and gaseous states disappears. If the equation of state is represented in a reduced form, i.e., in dimensionless parameters p/pc, ν/νc, T/Tc, then the equations of state of various substances differ only slightly (the so-called law of corresponding states) within the limits of a definite group (simple liquids, alkali metals) over a reasonably large near-the-critical area.
The weak dependence of liquid structure on temperature at constant density is the decisive factor in creating models of equations of state for a liquid.
Molten metals, ionic systems and dielectrics, having substantially different attractive forces, and this testifies to the small role these forces play in forming the equilibrium properties of a liquid. These facts reveal the leading contribution of repulsive potential, while attractive forces and accounting for the temperature effects in a liquid bring about only minor corrections. A simple potential of interaction of solid spheres with radius a is widely used as a model for the description of repulsive forces in the system:

which characterizes the limit state of a highly-compressed and hot liquid. The model for hard spheres, being the simplest nontrivial model of the equation of state of a liquid, is widely used in Integral Equations, numerical Monte-Carlo methods and in molecular dynamics. Using different methods, the calculation of a system of particles with the potential of solid spheres is made over the entire range of a liquid state. This can be represented with high accuracy by the Pade analytic Carnahon-Starling approximation

which has a free parameter ν = 4πa3n/3 (n is the number density of the particles) that characterizes the degree of solid sphere packing. It must be noted that numerical calculations of the potential of solid spheres, with density approximating the dense packing of spheres ν ≤ 1, have shown a tendency towards the appearance of a long-range order in the system which is associated with crystallization.
Considerable progress in describing the thermodynamic properties of a liquid phase have been attained with the help of a soft sphere model. This model uses a power potential of repulsion

which is also the basis for carrying out numerical calculations by the Monte-Carlo method and by the molecular dynamics method. The resulting calculations of the equation of state of soft spheres is described for any 4 ≤ n ≤ 12 by the approximating formula

where η = a3n/√2 and ε and n are the additional free parameters of the model. The power potential of repulsion, like the potential of hard spheres, brings about crystallization—whose characteristics depend strongly on the value of n.
A modified van der Waals’ model based on the combination of an approximate formula for the potential of soft spheres and an approximated average field considerably improves the description of the thermodynamic properties of a dense gaseous phase and a liquid. Greater accuracy in describing experimental data is achieved by using the results of calculations for the thermodynamics of a system of soft spheres and introducing adjusting parameters into the average field model. The modified van der Waals’ equations of state can be equally useful for calculating the thermodynamic properties of liquids and dense gaseous phase. They have asymptotics of an ideal gas, which makes it possible to use them successfully for expressing a wide-range of semiempirical equations of state. These models can also define the equilibrium liquid-vapor line and the critical point. But as a whole, they are quantitative and do not give a detailed description of the fine characteristics of a liquid; in particular, the dual correlation function g(r) which defines all equilibrium properties of a liquid phase for a given potential of interparticle interaction φ(r).
The density of a liquid near the melting point curve differs slightly (by 10–20%) from that of a crystal, which explains the success of equations of state for describing solid-state models. The notion of liquid molecules localizing in a small area in space under the action of an average field of neighbors forms the basis for the lattice model, or for the unconfined space theory. Such an approach, however, is applied only for a strongly-compressed liquid at low temperatures when kinetic energy forms a small fraction of total energy, a condition which, in general, does not correspond to a liquid state. A combination of solid-state and gaseous notions brings about a multistructural model, according to which an actual liquid is an equilibrium mixture of gaseous and solid bodies, whose properties can be described by a dual distribution function. Similar notions are used in a cell model where the liquid is divided into molecular cells, which are filled depending on thermodynamic conditions. Such models describe only schematically the thermodynamic properties of a real liquid.
Models of hard and soft spheres properly take into account the basic qualitative characteristics of multiparticle interactions in a dense liquid. This makes it possible to use them as a zero approximation model in calculating equations of state. The details of real potential of interparticle interaction are carried out within the framework of the thermodynamic perturbation theory, the chaotic phase approximation or on the basis of the variational method of perturbation theory. The variational method is most convenient for calculating the thermodynamics of particular liquids over a wide range of parameters and is especially effective in describing the properties of a dense liquid phase. Gibb’s-Bogolyubov’s inequality is used in finding the free energy F(V, T) of a system of particles with an arbitrary potential of interaction φ(r)
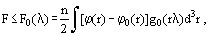
where F0 and g0 are the free energy and a dual correlation function of the original system with interparticle interaction potential φ0(k), respectively. The equation of state of an arbitrary system is defined by averaging the perturbation over the structural characteristics of an initial approximation and further minimizing a free parameter of this approximation λ according to (∂F/∂λ)V,T = 0. It is good practice to take a system of solid spheres as the zero approximation, for there are analytical expressions defining the free energy and dual correlation functions. This allows the explicit calculation of the thermodynamic characteristics of an arbitrary potential of the interparticle interaction. The radius of solid spheres is, in this case, the minimizing parameter λ. But at large densities, use of the solid spheres system as the initial one brings about essential errors, caused by the strong repulsion of particles at small distances. The choice, as the zero approximation model, of “soft” power potential φ(r) = C/r12 is usually favored here. Use of the soft and solid spheres model for the original approximation results in equations of state of inert liquids or molecular gases and simple metals that are of high accuracy.
The development of the variational method of perturbation theory for calculating the equation of state of liquid metals has led to the use of the results obtained from the Monte-Carlo method for a single-component plasma as the zero approximation. The model of a single-component plasma is based on numerical simulation of a simple system of point-like particles with Coulomb interaction, located, for its stability, on a homogeneous background of a balance charge with an opposite sign. In this case, there exists an analytical expression for F0, with the Coulomb nonideality parameter Γ = e/kTrws as the minimizing quantity, and the average interparticle distance rws = (3/4πn)1/3. Results of calculating the equations of state have shown that for liquids with slight repulsion (alkali metals, solt melts, alloys), a single-component plasma model is best suited. It should be noted that the final expression for the equation of state of liquid metals—in addition to the configurational term determined by the variational method, the expression for energy—must include terms according to the Coulomb interaction energy and the kinetic energy of particles of the system, whose total value at high temperatures becomes significant. In simple liquids, the same holds true when accounting for electron excitation at high temperatures.
Additional information on equations of state are given in the article PVT Relationships.
To form equations of state for a solid body, phonon and electron components are distinguished and are considered separately to a large degree. A distinguishing feature of solid bodies is their atoms which oscillate about the nodes of a crystal lattice. A quasi-harmonic model of a solid body is based on the representation of a crystal lattice in the form of 3N oscillators. The most widespread interpolation of the Debye equation for a solid body has been obtained in the approximation of a sound spectrum of heat variations, which yields the following expression for heat components of energy and pressure:

where 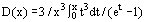 is the Debye function, θD is the Debye temperature dependent on volume and γ = − d lnθD/d lnV is the Grüneisen coefficient. The Debye function D(θD/T) ensures in this case a low-temperature cν ∞ T3 and a high-temperature
is the Debye function, θD is the Debye temperature dependent on volume and γ = − d lnθD/d lnV is the Grüneisen coefficient. The Debye function D(θD/T) ensures in this case a low-temperature cν ∞ T3 and a high-temperature 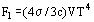 asymptotes for the heat capacity of a lattice. In the Debye model, the solid body is taken to be homogeneous and isotropic, thermal excitations of a crystal boil down to sound waves. External pressure in this case only influences the boundary temperature, while the spectrum itself remains harmonic.
asymptotes for the heat capacity of a lattice. In the Debye model, the solid body is taken to be homogeneous and isotropic, thermal excitations of a crystal boil down to sound waves. External pressure in this case only influences the boundary temperature, while the spectrum itself remains harmonic.
Calculations for an electron spectrum are usually made within the framework of the zone model. Here the crystal presents an ideal periodic structure with immobile cores in the cell nodes and each electron moves in a periodic self-consistent potential, which allows expressing the wave function of the whole system through single-electron Bloch’s functions. The distinction between the models of equations of state reduces to the methods of calculating single-electron functions within the elementary atomic cells, and to the methods of joining the conditions at their boundaries. In the widely-used Wigner-Seitz model of spherical cells, a complete self-consistent problem is replaced by the solution of the Chariri-Fock equation for a single cell with Bloch’s boundary conditions. In doing this, zones of allowed states for electrons arise. The association of pressure with wave functions on the cell boundary makes it possible to determine the equation of state of the system.
Under normal conditions, the approach of spherical cells is too schematic; besides, a considerable contribution is made by the effects of exchange and correlation interactions. For this reason, methods of adjoined flat or spherical waves, linear orbitals, etc., have been developed which accounted for the real shape of an elementary crystal cell. Exchange-correlation effects are accounted for in local density approximation.
A pseudo-potential model of a solid body is based on a true potential interaction of an electron with multicharge ion onto a simplified, single-particle. A consecutive theory of pseudo-potentials accounting for two-, three-, etc., ionic interaction through conduction electrons is a rigorous expansion into a series in terms of the parameter of electron-ion interaction. The application of a pseudo-potential approach at high degrees of compression is limited by the effect of ion core overlapping.
The increase in temperature brings about the appearance—both in gas and in liquid—of free charges, with the long-range character of the Coulomb interaction making significant its contribution into the equation of state over a large area of thermodynamic parameters. In a rarefied plasma, a separate description of states of discrete and continuous spectra—which define the internal structure of atoms and ions as well as the character of the behavior of free electrons—is practiced. Such an approximation forms the basis of a “chemical” model of plasma in which the number of particles of {Nj} kind is determined from the conditions of chemical equilibrium

where μj is the chemical potential of particles, and all the hypotheses specifying structure and interaction are involved in the expression for total free energy. This expression includes the kinetic energy of ions and electrons of a continuous spectrum, the contribution of the discrete spectrum and various corrections for interparticle interaction. At extremely high temperatures of the plasma or with strong rarefaction, the energy contribution of a photon gas F1 = (4σ3c)VT4 (σ is the Stefan-Boltzmann constant and c is the velocity of light).
REFERENCES
Godwall, B. K., Sikka, S. K., and Chidambaram, R. (1983) Equation of state theories of condensed matter up to about 10 TPa Phys. Rep. 102. DOI: 10.1016/0370-1573(83)90014-5
Ross, M. 1985. Matter under extreme conditions of temperature and pressure Rep. Prog. Phys., 48.
参考文献
- Godwall, B. K., Sikka, S. K., and Chidambaram, R. (1983) Equation of state theories of condensed matter up to about 10 TPa Phys. Rep. 102. DOI: 10.1016/0370-1573(83)90014-5
- Ross, M. 1985. Matter under extreme conditions of temperature and pressure Rep. Prog. Phys., 48. DOI: 10.1088/0034-4885/48/1/001