Irreversible thermodynamics is a division of physics which studies the general regularities in transport phenomena (heat transfer, mass transfer, etc.) and their relaxation (transition from nonequilibrium systems to the thermodynamical equilibrium state). It is possible to use for this purpose, as in reversible thermodynamics (thermostatics), phenomenological approaches based on the generalization of experimental facts and statistical physics methods which establish the bonds between molecular models (microscopical structure, properties of molecules, intermolecular interaction) and macroscopical substance behavior.
The starting points of irreversible thermodynamics are the first and second laws of thermodynamics in local formulation. In the general case, the nonequilibrium continuum is a moving v-component mixture of substances A1, A2, ... Av, among which r chemical reactions run simultaneously

It is described by the fields of temperature T(t, r), mass concentrations of components ρi(t, r) and average mass flow speed v(t, r). The local density of system ρ(t, r) and mass fractions of components хi (t, r) are defined by the relations

The conservation equation of mixture mass and balance equations of component masses in the local coordinate system, moving with the mass center speed v(t, r), can be written as:

Here, ji is the diffusion flow density of the ith component,

and Rk is the rate of the kth chemical reaction. The balance equation of specific internal energy u(t, r) has the following form:
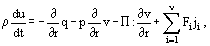
where q is the heat flow density, p is pressure and Π is the viscosity part of pressure tensor

∂r/∂r is the speed deformation tensor, Fi is the mass force exerted on the ith component particles from the external fields. Equation (5) is the local formulation of the first law of thermodynamics for a nonequlibrium open system.
The second law of thermodynamics in the form of a local entropy balance is deduced by substituting Eqs. (3) and (5) with the thermodynamical relationship

describing the variation of local specific entropy s(t, r) with time interval dt in the center mass coordinate system of the continuum. In Eq. (6), v = 1/ρ is the specific volume, and μi(t, r) is the locai specific chemical potential of ith component. In so far as du in Eq. (6) contains the dissipative contributions (the two last terms on the right side of Eq. (5)); ds includes both the "equilibrium" component dse and the nonnegative term dsi ≥ 0; and conditioned by irreversibility:
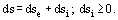
The local entropy balance equation follows from Eq. (6):
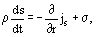
where

is entropy flow density, and

is entropy production. In Eq. (9),
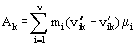
is the affinity of chemical reaction (Eq. (1)). Equation (9) has the structure
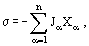
where Jα are the flows (Ji, g, Π, Rk) and Xα are the matching thermodynamic forces. The second law of thermodynamics is reduced to

Equality in Eq. (11) corresponds to the thermodynamical equilibrium state—full or local, and nonequal—for the local entropy growth in irreversible processes. From the second law of thermodynamics (Eq. (11)) can be inferred the fact that flows in Eqs. (9) and (10) are in opposite direction to thermodynamic forces, JαXα ≤ 0 and they disappear after the transition to thermodynamic equilibrium. Practice fully confirms these deductions.
In accordance with experience, flows and thermodynamic forces are bound by transfer laws Jα = F(Xj, . .., Xn); 1 ≤ α ≤ n; moreover if thermodynamic forces are absent, the flows are also equal to zero. The most simple relationship is linear,

The assumption about linearity of transport laws is a basis of linear, irreversible thermodynamics. Quantities Lαβ are called kinetic (transport) coefficients. They are properties of the system considered; it means they don't depend on thermodynamic forces and flows, but are functions only of the state parameters (temperature, pressure, mixture composition). Phenomenological irreversible thermodynamics neither establishes these dependencies nor indicates suitable limits to linear transport laws. However, it is possible to formulate a number of general statements about the structure and properties of the kinetic coefficient matrix Lαβ.
From Eqs. (9) and (10), it is clear that flows and thermodynamic forces have different tensor dimensions. Diffusion flows ji and diffusion thermodynamic forces

are vectors, as well as heat flow g and the related thermodynamic force (l/T)∂T/∂r. Reaction rates Rκ and affinities Aκ are scalars. Some further scalar couples (J,X) are obtained from the last but one term in Eq. (9) the normal and tangential viscosity tension contributions are given by π = (Πxx + Πyy + Πzz)/3;  = Π − π1, and it also follows that
= Π − π1, and it also follows that

(S is the shift rate tensor, ∂/∂r; ≡ div). Thus π and (l/T)(∂/∂r) · v are the scalar flow and the thermodynamic force, respectively; flow  and force S/T are second-range tensors. According to the Courier principle, only flows and thermodynamic forces with the same tensor dimensions may be related using linear correlations. Any kinetic coefficient Lαβ, relating a flow Jα and a thermodynamic force Xβ with different tensor dimensions is identically equal to zero. The permissible linear correlations between flows and thermodynamic forces occurring in nonequilibrium, multicomponent reacting moving fluid (Eqs. (3−9)) are terms limited to
and force S/T are second-range tensors. According to the Courier principle, only flows and thermodynamic forces with the same tensor dimensions may be related using linear correlations. Any kinetic coefficient Lαβ, relating a flow Jα and a thermodynamic force Xβ with different tensor dimensions is identically equal to zero. The permissible linear correlations between flows and thermodynamic forces occurring in nonequilibrium, multicomponent reacting moving fluid (Eqs. (3−9)) are terms limited to





The correlations (Eq. (12) and Eqs. (13−17)) predict not only direct (Jα = LαXα) but "overcrossed" irreversible processes when the flow of a certain physical characteristic is implemented by other natural, "nonrelated" thermodynamic forces (Jα = LαβXβ; α ≠ β). Thus Eq. (13), the generalized Ficks law, describes diffusion in a multicomponent mixture when diffusion flow ji of component i is caused by the diffusion thermodynamic forces di (in particular, by concentration gradients ∂ρj/∂r) of other components as well as temperature gradient. The quantities Lii and Liq represent the multicomponent diffusion and thermal diffusion coefficients.
The generalized Fourier law (Eq. (14)) describes a rise of heat flow at the expense of temperature gradient ∂T/∂r (the transport coefficient of this direct process is related to the ordinary thermal conductivity λ = Lqq/T2), and by diffusion thermodynamic forces di. The latter process is called the diffusion thermo effect. It is in mutual overcross with thermal diffusion. The appearance of tangential viscous tensions, which is described by the generalized Newton-Stokes law (Eq. (15)), is a direct process (dynamic viscosity coefficient η = [1/2] [L/T]) and the overcross processes are absent here. Yet normal viscous tensions (at is clear from Eq. (16)) can arise both because of the direct process—which stipulates volume expansion of the flow and is formed by the volume viscosity coefficient κ = Lpp/T—and the overcross processes arising from nonequilibrium chemical reactions.
All the above irreversible processes have been observed in practice and are really linear until the fluid in which they proceed can be considered as a continuum.
It is impossible to say the same about the conditions of Eq. (17). The linearity of conditions between chemical reaction rates and their affinities is broken already by small deflections from chemical equilibrium Ak = 0, Rk = 0.
Uniting Eqs. (11) and (12), it is possible to present entropy production in quadratic form

According to Eq. (11), it is positively determined. Hence it follows that all the diagonal elements of the kinetic coefficients matrix forming direct irreversible processes are positive, Lαα ≥ 0; all its principal minors are positive too. Moreover, important information on the structure of this matrix is derived from Onzager mutuality correlations, which establish equality of kinetic coefficients for overcrossed, irreversible processes:
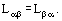
The derivation of accurate formulas for kinetic coefficient calculation from molecular data, as well as molecular kinetic justification of the second law of thermodynamics for nonequilibrium systems in the form of Eqs. (7)−(11), is feasible now only for dilute gases by applying the Boltzman kinetic equation and its modifications. Attempts to describe dense nonequilibrium system behavior proceeding from reversible, equations of molecular motion meets with problems that cannot be completely overcome. Together with the kinetic equations method, the nonequilibrium statistical operator method and related approaches of linear reaction theory can be successfully employed.
In certain cases, the introduction of some critical nonequilibrity degree can suddenly increase an open system's regularity, which is followed by dissipative structure formation. Known examples are: the occurrence of Benard cells in a viscous liquid layer, heated from below in gravitational field, when the heat flux exceeds a critical value; the appearance of layers in a gas discharge column with the introduction of a critical current density; and so on. A richer variety of dissipative structures—both space and temporary—are found in nonlinear, noneqilibrium systems (for example, the Belousov—Zabotinski reaction). A comparatively new and swiftly-developing branch of irreversible thermodynamics is devoted to the study of the organization of phenomena in strong nonequilibrium, nonlinear open systems.
REFERENCES
de Groot, S. R. and Mazur, P. (1962) Nonequilibrium Thennodynamics. North-Holland Publ. Co. Amsterdam.
Glansdorf, P. and Prigogine, I. (1970) Thermodynamic Theory of Structure, Stability and Fluctuations. A division of John Wiley & Son. London/New York/Sidney/Toronto.
Nicolis, G. and Prigogine, I. (1976) Self-organization in Nonequilibrium Systems. A. Wiley Interscience Publ. New York/London/Sidney/Toronto.
Les références
- de Groot, S. R. and Mazur, P. (1962) Nonequilibrium Thennodynamics. North-Holland Publ. Co. Amsterdam.
- Glansdorf, P. and Prigogine, I. (1970) Thermodynamic Theory of Structure, Stability and Fluctuations. A division of John Wiley & Son. London/New York/Sidney/Toronto. DOI: 10.1126/science.176.4042.1410
- Nicolis, G. and Prigogine, I. (1976) Self-organization in Nonequilibrium Systems. A. Wiley Interscience Publ. New York/London/Sidney/Toronto. DOI: 10.1080/03081077808960692