Multicomponent systems are the mixtures (the solutions) which consist of several pure (individual) substances (components). The initial amount each of the substances in the ν-component system may be represented in terms of the component masses mi (1 ≤ i ≤ ν). Another way, which is widely used in thermodynamics, is to represent the system composition in terms of the number of moles of a component, Ni = mi/Mi, where Mi is the molecular mass of the component.
The thermodynamic behavior of the v-component system is described by the First Law and the Second Law of thermodynamics. If a system under consideration is an open one, then, not only energy exchange in the form of work or heat may take place but also mass exchange. When there are no external force fields the joint equation of the First and the Second Laws may be written in the form:

where U, S and V are the total internal energy, entropy and volume of the system; μi is a chemical potential. An equality sign is valid in Eq. (1) for equilibrium processes or, in other words, for reversible or quasistatic processes; an unequality sign relates to the case when nonstatic processes or dissipative effects take place.
Equation (1) defines as the internal energy U as a thermodynarnic potential with regards to independent variable S, V, N1, . . . Nv. If the set of the independent variables is not convenient to use. then after the Lejandre transformation of Eq. (1) it is possible to introduce the other thermodynamic potentials such as an enthalpy H(S, p, N1, . . ., Nv) = U + pV; a Helmholtz energy A(T, V, N1, . . ., Nv) = U - TS; a Gibbs energy G(T,p,N1 ..., Nv) = H - TS. For the last case Eq. (1) may be rearranged into the form

which allows the calculation of any thermodynamic function for a multicomponent system under equilibrium. For this case the Gibbs energy should be known as a function of its own independent variables T, p, N1 ... , Nv, which is more convenient for practical applications. For equilibrium conditions it follows from Eq. (2) that:


A chemical potential μi of the component i may be calculated by differentiating any of the similar thermodynamic potentials, with respect to the number of moles of the chosen component:

Also from Eq. (1) and (2), exact correlations may be derived (the Maxwell equations), which are the joint thermodynamic parameters of the multicomponent system. These correlations may be used for describing the behavior of the system under consideration if limited experimental data is available.
To describe the composition of a multicomponent system, concentrations of the components are used: in terms of mass ρi = mi/V as well as of moles
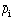 = Ni/V, which define the content of the component per unit volume. Fractions of the components are also commonly used: thus mass fraction χi = mi/m and mole fraction
= Ni/V, which define the content of the component per unit volume. Fractions of the components are also commonly used: thus mass fraction χi = mi/m and mole fraction
 = Ni/N, where m and N are the total mass and the total number of moles in the system, respectively. System compositions in terms of molar quantities
= Ni/N, where m and N are the total mass and the total number of moles in the system, respectively. System compositions in terms of molar quantities
 or
or
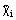 are more convenient to use in chemical thermodynamics or in the thermodynamics of solutions.
are more convenient to use in chemical thermodynamics or in the thermodynamics of solutions.
It is easy to calculate the mass compositions when the mole composition values are known:

where an equivalent molecular mass of the mixture M is defined as

Extensive thermodynamic functions such as V, H, G, S, etc. which appear in Eqs. (1)-(3) depend not only on thermodynamic parameters of the system but on the amount of the substance included in the system. As such, the extensive values can not be considered thermodynamic properties. On the contrary, the intensive thermodynamic functions—such as specific volume v, enthalpy h, Gibbs energy g, etc.—which have been obtained dividing the extensive functions by the total mass or the total number of moles the system, are exactly defined by the same parameters which characterize a thermodynamic state of the system. In this sense, the intensive functions may be considered as thermodynamic properties of the system. Below we will use molar thermodynamic values defined for any extensive function Ψ in the form

To obtain the corresponding specific (mass-based) values, the molar functions of the form (6) have to be divided by molecular mass of the mixture defined by Eq. (5).
By definition the chemical potential mi appearing in Eqs. (3) and (4) is an intensive thermodynamic function as are other functions of the type

which used to be called partial thermodynamic functions: for example, partial volume
 and entropy
and entropy
 . It follows from Eq. (3) that the chemical potential μi is equal to the partial Gibbs's energy
. It follows from Eq. (3) that the chemical potential μi is equal to the partial Gibbs's energy
 of the ith component.
of the ith component.
Any thermodynamic functions, whether extensive or specific, may be determined according to the additive rule:
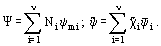
Both molar and specific thermodynamic functions of the form defined in Eq. (6) and partial functions defined as in Eq. (7) satisfy the same thermodynamical correlations as the corresponding extensive functions. The correlations between the partial functions and the mixture composition given as
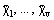 may be obtained from the Gibbs-Duhem equation
may be obtained from the Gibbs-Duhem equation

Any variations of the partial functions caused by a small change of the mixture composition satisfy Eq. (9) if T and p stay constant.
Equation (8) does not imply that properties of any multicomponent mixture may be calculated if these properties were known for the components. This is possible only for the ideal mixtures (solutions). In such cases, the partial functions with exception of these containing entropy (i.e., volume, internal energy, enthalpy, heat capacity, etc.) will be equal to the corresponding specific functions calculated for the pure components at given T and p (Amagat's Law). In general this may be written as:

(It is implied that the aggregate state should be the same for each of the components and the mixture.) For an ideal mixture, the partial entropy si as well Gibbs energy
 contains terms additional to those indicated in Eq. (10):
contains terms additional to those indicated in Eq. (10):


These terms take into account the entropy increase due to the irreversible mixing of the components:

Equation (12) is a form of Raoult's Law.
For mixtures whose components are perfect gases only, the Gibbs energy of the components gi(T,p) in Eq. (12) may be calculated as

where p0 = 101325 Pa is the standard atmospheric pressure and
 (T) is the standard Gibbs energy of the pure substances.
(T) is the standard Gibbs energy of the pure substances.
The perfect gas mixtures obey not only Amagat's and Raoult's Laws but also Dalton's Law which gives the correlations:

Real mixtures do not obey the Amagat's and Raoult's Laws. The deviations from the ideal solution behavior are described by the excess thermodynamic functions

The excess volume υE enthalpy hE, entropy sE define the volume, heat and entropy of mixing effects accordingly. As a result of the mixing effects, the system changes volume and a heat of mixing may may need to be added or removed from the system, even at constant p and T. Also the entropy change may be greater or less than Dsm calculated from Eq. (13). These effects not observed in ideal mixtures, but they are important in a qualitative and quantitative sense to describe the behavior of real mixtures. The effects are due to molecular interactions, which are essentially different in pure substances and in mixtures where the interactions of different kinds of molecules take place.
Activity coefficients (γi) are commonly used in the thermodynamics of solutions instead of excess partial Gibbs energies
 . The two quantities are related by the defining equation:
. The two quantities are related by the defining equation:

If the mixture is an ideal then for all the components γi = 1.
From Eqs. (12), (16) and (17) it is possible to relate
 to activity coefficient:
to activity coefficient:

The excess Gibbs energy
 is also related to activity coefficient by:
is also related to activity coefficient by:

If the dependence
 on
on
 , ...
, ...
 known, then the activity coefficients γi for all the components may be calculated.
known, then the activity coefficients γi for all the components may be calculated.
The volumetric, heat and the other mixing effects may be deduced from Eq. (19) if the equations similar to (3) are used:

Similar equations may be used to calculate from Eq. (17) any of the excess partial thermodynamic functions of the components:

In order to describe thermodynamic properties of real gases mixtures, equations of state given as functions of pressure on temperature, molar density and mixtures composition are used:

(if a mixture includes ideal gases only, then z = 1). Statistical mechanics allows the derivation of the exact function (20) having correlated it with a potential energy of the molecular interactions. Such equations are valid for gases of moderate density. For high-density gases and liquids, there are many empirical equations of state. Many of them represent the modifications of the famous van der Waals Equation. Such equations may often describe both the gaseous and the liquid phases of pure substances and their mixtures. If the equation of state (20) is available, then it is possible to calculate any of the thermodynamic functions of the mixture in terms of the variables T,
 ,
,
 , ...
, ...
 . For example,
. For example,

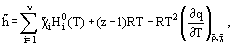
etc., where
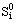 =
=
 ,
,
 =
=
 are the standard entropy and enthalpy, and
are the standard entropy and enthalpy, and

In many cases it is more convenient to use, instead of a chemical potential μi =
 , a Fugacity fi of the i-component. Based on Eqs. (12), (14), and (15), a fugacity may be introduced as an analog of a partial pressure pi (15):
, a Fugacity fi of the i-component. Based on Eqs. (12), (14), and (15), a fugacity may be introduced as an analog of a partial pressure pi (15):

For the mixture comprising the perfect gases the fugacity of a component equals to its partial pressure:

If a pure substance fugacity is defined in a manner similar to (23) as

where
 is a molar Gibbs energy of the pure i-substance, then according to the definition (18) it is possible to obtain a relationship which links the fugacities (23) and (24) in the real mixture to the activity coefficients (17):
is a molar Gibbs energy of the pure i-substance, then according to the definition (18) it is possible to obtain a relationship which links the fugacities (23) and (24) in the real mixture to the activity coefficients (17):

Taking into account (25) Raoult's Law (12) may be rearranged into the form of the Lewis Rule:
 .
.
The fugacity coefficient φi = fi/p
 can be evaluated from equation of state (20) taking account of Eq. (21):
can be evaluated from equation of state (20) taking account of Eq. (21):

In order to calculate the thermodynamic functions (21), (22), (26) of the mixture at given T,p,
 , ... ,
, ... ,
 , it is necessary to solve (as a rule numerically) the nonlinear Equation (20) with regards to the mixture density
, it is necessary to solve (as a rule numerically) the nonlinear Equation (20) with regards to the mixture density
 using a defined route in the application of the correlations which are to be used for the purpose, if the equation of state (20) describes both a gas and a liquid phase, then in the vapor-liquid phase there exist several routes. The minimal of them relates to the gaseous and the maximal one to the liquid phase, all the other routes have no physical sense and could describe none of the stable states of the system.
using a defined route in the application of the correlations which are to be used for the purpose, if the equation of state (20) describes both a gas and a liquid phase, then in the vapor-liquid phase there exist several routes. The minimal of them relates to the gaseous and the maximal one to the liquid phase, all the other routes have no physical sense and could describe none of the stable states of the system.
In summary, there are two methods of describing the thermodynamic behavior of multi-component systems: one of them is based on excess functions and another on the equation of state. Both of them are equivalent, but each has its own advantages. The first one has been found best for mixtures all the components of which, at the given p and T, are in the same aggregate state as the mixture one. The method is difficult to use in describing the phase equilibrium and critical phenomena in solutions. The second method, however, is invalid in describing solid solutions and melting processes. Even the most modern equations of state are less accurate than the experimental data they aim to describe.
REFERENCES
Prigogine, I. and Defay, R. (1954) Chemical Thermodynamics. Longmans, London.
Reid, R. C, Prausnitz, J. M. and Poling, B. E. (1987) The Properties of Gases and Liquids. McGraw-Hill Book Co.
Walas, S. M. (1985) Phase Equilibrium in Chemical Engineering. Butterworth Publishers.
References
- Prigogine, I. and Defay, R. (1954) Chemical Thermodynamics. Longmans, London.
- Reid, R. C, Prausnitz, J. M. and Poling, B. E. (1987) The Properties of Gases and Liquids. McGraw-Hill Book Co.
- Walas, S. M. (1985) Phase Equilibrium in Chemical Engineering. Butterworth Publishers.