This article deals with the prediction of corrosion inside carbon steel pipes carrying unprocessed hydrocarbons and in pipes used for injection of water into reservoirs (the injected water will maintain the pressure in the reservoir).
Several forms of corrosion may occur in such systems. Depending on the form of corrosion, the predictions may result in: a) "yes" or "no" rating of a material; or b) a predicted corrosion depth after a certain length of service. An example for a) is if the partial pressure of H2S is larger than 0.003 bar and the pH and temperature are smaller than certain magnitudes, stress corrosion cracking of carbon steel may occur. A carbon steel with a specified heat treatment and hardness will then obtain a "yes" rating.
For b), an example is general CO2 corrosion. This form may allow operation in an environment which induces significant corrosion of the carbon steel. The expected lifetime is now compared with a predicted cumulative corrosion depth. In order to preveni reduction of the operating pressure in the pipe, the predicted corrosion depth must be equal to or smaller than the corrosion allowance. i.e., the extra thickness of pipe which may be consumed by corrosion.
Prediction of corrosion depth in carbon steel relies largely or experiments. Based on experience, present-day mechanistic models do not allow estimates of corrosion depth outside the parameter range covered in experiments. One reason is the inability to predict how protective a corrosion product will be – i.e., its porosity and strength – without experiments. No mechanistic model can simulate how a film-forming inhibitor will influence the corrosion process. If a film-forming inhibitor is added to water, the inhibitor may, for example, modify the characteristics of the corrosion product in a manner specific for the given inhibitor. It is even possible that certain inhibitors may prevent formation of this product. Further partial filming may induce pitting. Thus, mechanistic models are limited to the estimation of how, for example, certain additives to the water can modify the pH and hence the corrosivity of a fluid.
Below, two important forms of corrosion which can be predicted by mechanistic models will be demonstrated. These are:
Production systems – CO2 corrosion of carbon steel
Injection systems – O2/Cl2 corrosion of carbon steel
The laminar flamelet model, with assumed probability density functions (PDFs) which are functions of the reaction progress variable and the stretch rate, appears to be valid over a wide range. It has been used successfully to predict lift-off heights of turbulent diffusion flames, the combustion field in premixed swirling flow, and flame blow-off.
Corrosion of carbon steel in a CO2 environment (production of hydrocarbons) and of such steel in an O2/Cl2 environment may serve as examples of corrosion processes when mass transfer of the corrosion species through electrolytes may partly or completely control the corrosion rates.
In practice, a model is used for prediction if the corrosivity of the environment allows the use of carbon-steel when an effective inhibitor is applied. An effective inhibitor should reduce the general corrosion rates by 80-90% and should not induce pitting in the steel.
The next step is to verify the corrosivity and select the inhibitor by realistic experiments, i.e., in high-pressure hydrocarbon gas/liquid flow loop [Gramme (1994)]. Later, inhibitor effectiveness should be verified during production for actual field application.
Here, however, only the description of a mechanistic model will be given. The model described by Nordgaard and Soendvedt (1994) will serve as a basis.
The corrosion rate of steel in CO2 environment appears to be mainly controlled by cathodic reaction, which most probably is the hydrogen evolution reaction [Smith and Rothmann (1979)].
There are discrepancies in the literature as to the species reacting at the electrode to form hydrogen atoms. The applied cathodic reaction mechanism is based on the work by de Waard and Milliams (1975) who suggested that the undissociated acid plays a catalytic role in the cathodic process
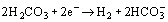

with Eq. (1) as the rate-determining step. The anodic reaction is as follows:
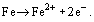
From these reactions, it can be seen that H2CO3 is consumed and Fe2+ and HCO3− are generated by the reaction.
According to the electrochemical theory of corrosion, the corrosion process takes place at a mixed potential Ecorr. It is assumed that the Bulter-Volmer expression can be applied to the kinetics of system reactions [Newman (1973)]. Based on experimental data from de Waard and Milliams, the reactions flux can then be given by
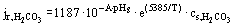


where
| jr = reaction flux at the surface; |
| A = constant describing the pH dependence of surface reaction; |
| PH6 = pH at surface; |
| T = bulk fluid temperature; and |
| Cs,i= concentration of species i at the surface. |
The thermodynamic model is based on a work by Liu and High (1992). The bulk concentrations are calculated, assuming that the solution is saturated with CO2 and chemical equilibrium is established for the system, by

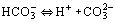

If the system satisfies the equation involving the solubility product criterion, an additional equation is required,
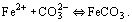
At present, the species involved are H2CO3, H+, Fe2+, Na+, OH−, HCO3− and CO32−, and electroneutrality can be expressed as:

The equilibrium constants are taken from the work by Edwards et al. (1978). The concentration of H2CO3 can be obtained from Henry's law

where PCO2 is the partial CO2 pressure and HCO2 is the Henry constant.
In order to calculate the bulk concentrations, the activity coefficients need to be evaluated. The calculations are the same as in SOLMINEQ88 [Kharaka et al. (1988)].
General formulation. The corrosive gas, CO2, will dissolve into the liquid phase, H2CO3. H2CO3 will disassociate into its corresponding ions, HCO3− and CO32−. Ions and H2CO3 are transported to the pipe wall where corrosion will occur. In the corrosion process, surface electrochemical reactions will set up an electric field. A feature of ions transfer in a moving solution that differentiates it from the transfer of dissolved neutral particles is the fact that the motion of ions is affected by the electric field in the solution. Ion transfer in the solution is produced by convective diffusion and by the migration of ions in the electric field. Convective diffusion in the solution is governed by two quite different mechanisms. Molecular diffusion is the mechanism of transport of species to the electrode as a result of concentration differences causing a diffusional flux

where Dmdi is the molecular diffusion coefficient for species i and ci is the concentration of species i.  ci is given by
ci is given by

In a moving liquid, the solute is entrained and transported by the flowing stream, causing a convective flux
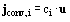
where the velocity field, u, is defined as:
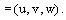
The migration of i-type ions can be written in the form

where R is the universal gas constant, F is the Faraday constant and zi is the valency of ion i. The electric field, E, is given by

where φ is the electric potential. The total flux of particles of the species i in the moving medium, ji is equal to

Diffusion in turbulent flow in a pipe Levich (1962) has shown that turbulent flow has a four-layered structure. Far from surface, there is a zone of developed turbulence in which the concentration remains constant. Closer to the surface, in the turbulent boundary layer, both average velocity and average concentration decreases slowly according to a logarithmic law. In this zone, neither molecular viscosity nor diffusion play a noticeable part. Both momentum and matter are transferred by means of turbulence eddies. Still closer to the wall, in the viscous sublayer, turbulence eddies become so small that the momentum transferred by molecular viscosity exceeds that transferred by turbulence eddies. However, the molecular diffusion coefficient is 1,000 times smaller than kinematic viscosity, Sc >> 1, and the remaining turbulence eddies still transfer substantially more solute than molecular diffusion. The coefficient of turbulent diffusion (eddy diffusivity) in the viscous sublayer is proportional to y4 and decreases rapidly as the wall is approached. At a certain distance from the wall, δdl, eddy diffusivity must equal the coefficient of molecular diffusion.

where
| εD, i = eddy diffusivity; |
| γ = constant; |
| u* = friction velocity; |
| δdl = diffusion layer thickness; and |
| δdl = viscous sublayer thickness. |
The viscous sublayer thickness can be expressed as:
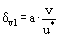
where ν is the kinematic viscosity. Solving for the diffusion layer thickness gives
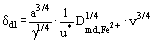
where γ = 1 and a =
 . The friction velocity is given by the following relation
. The friction velocity is given by the following relation

where
| τwi = wall shear stress; |
| ρ = liquid density; and |
| U∞ = pipe velocity. |
The skin-friction coefficient, Cf, (f = 4 · Cf) is a function of the Reynolds number and pipe roughness, k. An equation by Colebrook and White encompasses all flow regimes encountered in a Moody chart. This equation is:
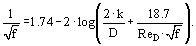
The molecular diffusion coefficients are obtained from dilute electrolyte theory

where λi is the limiting ionic conductance of species i in water at 298 K. In the innermost portion of the viscous sublayer at y < δdl, the molecular mechanism predominates over the turbulence mechanism. (See also Boundary Layer; Friction Factors for Single Phase Flow; Turbulent Flow.)
Flux and mass balance equations. At present, it is assumed that the diffusional layer provides all the resistance to mass transfer. Therefore, outside this layer, the concentration is uniform for all species and equal to the bulk concentration. A one-dimensional problem is considered. Equation (19) can then be simplified to yield

and the mass balance is reduced to

These equations involve an electrical potential term. Then, one more equation is necessary to completely specify the system. For a corrosion system, the assumption that the net current flow is zero (open-circuit condition) is always true [Levich (1962)]

From the latter, the distribution of the field in the solution can be derived

Boundary conditions. At pipe wall surface (y = 0):
The total reaction flux of species i in the moving medium in the diffusion layer is equal to

If the species are nonreactive at the wall, jr, i is zero.
The diffusion layer boundary (y = δ):
At the interface of the diffusional layer and the turbulent layer (outer part of the viscous sublayer), ci = cδ, i = cb, where cδ, i is the concentration of species i at the diffusion layer boundary and cb, i is the concentration of species i in the bulk.
The model needs bulk temperature, CO2 partial pressure, bulk velocity, bulk iron concentration and added sodium bicarbonate as input. Initial concentration values for all species in the bulk is calculated by assuming the activity coefficients to be unity. Based on these initial values, the actual concentrations are calculated. After calculating the physical properties (viscosity, density and diffusion coefficients), the diffusion layer and flow condition are calculated.
The diffusion layer is divided into grids, and initial values for all the grid points are calculated. The surface reaction fluxes are calculated for the given initial values. The grid point concentrations throughout the diffusion layer are calculated by a finite difference technique [Patankar (1980)].
Because ordinary differential equations are highly nonlinear and coupled, they are solved one by one. The specific equation generally converges in two or three iterations. When all the equations are solved, the concentration of the species are updated. As a convergence criteria, the total number of iterations are used. The program checks if convergence really occurred. If not, more iterations need to be performed.
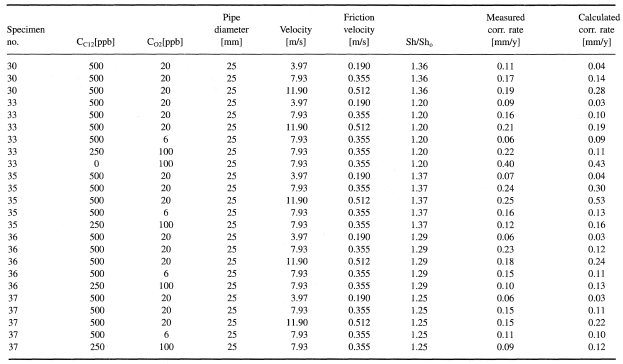
In order to test out the model, experimental results from Kjeller Sweet Corrosion have been used. These experiments are documented elsewhere [Dugstad and Videm (1990)]. Simulation results of the pH and the resulting corrosion rate along with the measured values are given in Table 1.

Experiments with temperatures up to 60°C have been applied. Above this value, the formation of protective layers is seen to lower the corrosion rate. The pH calculations agree very well with the values given for the experiments. The corrosion rate simulations are within 20% of the experimental results, which is regarded as a very good agreement.
Seawater used for injection into the reservoir is treated to avoid microbiological activity, corrosion and reservoir formation blockage. The need for both microbiological and corrosion control can be in conflict because chlorine, which is corrosive, is one of the chemicals used for microbiological control. Typical chlorine concentrations are 0.3-0.5 ppm [Hudgins (1971) and Mitchell (1978)].
Chlorine can be produced by electrochlorinators or added as hypochlorite. When chlorine is added to seawater, an equilibrium between chlorine (Cl2), hypochlorous acid (HOCl) and hypochlorite (OCl−) is established. At pH > 7.5, hypochlorite predominates whereas at pH 6.5, OCl− is present with considerable quantities of HOCl [Mitchell (1978)]. Another complicating factor is the presence of approximately 70 mg/l bromide in seawater. The bromide will have the effect of reducing the added hypochlorite to Cl− while Br− is oxidized to hypobromite (OBr−) [Gundersen et al. (1989)].
Several cathodic reactions can take place in chlorinated seawater.
Reduction of hypochlorite:

Or hypobromite:
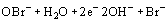
Reduction of oxygen:

Reactions (31), (32) and (33) are flow-dependent. Flow dependence will not only include the velocity, but also the geometry (i.e., obstacles with local enhanced mass transfer and wall shear stress). Corrosion rates have been measured in a simulated water-injection system where seawater has been chlorinated and deoxidized [Andersen and Soendvedt (1993)]. The influence of chlorine, oxygen, flow velocity and geometry have been investigated.
The following conclusions were made from the experiments:
Oxygen alone is more corrosive than in combination with chlorine.
In injection systems with chlorinated seawater, two types of layers are formed on the steel surface. In seawater with low oxygen concentrations, magnetite will be the corrosion product. It was also observed that the pipes were covered with a layer of organic matter when chlorine was added to seawater.
The corrosion rate seems to be controlled by mass transfer of oxygen and chlorine to a protective layer, and through this layer to the metal surface.
The corrosion rate is mainly controlled by the concentration of oxygen and chlorine and local mass transfer of corrosive species. The corrosion rates at flow disturbances are much larger than on smooth sections.
It is assumed that the corrosion rate is limited by transport of chlorine and oxygen through a protective layer. The flux density of each corrosive species through the diffusion boundary layer and through pores in the protective layer with thickness H and permeability Kp is given as follows:

where
| βm = local mass transfer coefficient to the protective layer; |
| H = height of layer made by both oxygen and chlorine; |
 = bulk concentration of species;
= bulk concentration of species; |
| Dmd, i = molecular diffusivity; and |
| Kp = porosity of layer. |
The total corrosion rate:

where
| K2 = corrosion rate corresponding with unit cathodic diffusion limited current; |
| z = valency change; and |
| F = Faraday constant. |
It can now be assumed that the thickness of the protective layer (H) is increased by mass transfer of each species to the layer (βm) and is decreased by wall shear (τW) from the liquid. In steady-state, this gives H equal to

where K is a constant.
Apparently the porosity of magnetite can be constant as described by Surman and Castle (1969) and Sanchez-Caldera (1984). The porosity is set equal to 0.3. For turbulent flow with a moderate flow normal to the wall, the wall shear is related to the mass transfer coefficient by:

When two pipes with different diameters have the same friction velocity, the wall shear on the pipe walls are identical. From the above relations, it follows that the height of the protective layer is given as

where K4 is a constant for a given liquid density;

where ρ is the density of the fluid.
A main variable, if the corrosion process is diffusion-controlled, is the local mass transfer distribution on a specific section of the piping. This local mass transfer can be very different from smooth-section magnitudes. This, in turn, governs the velocity dependence by enhanced transport to the protective layer and the equilibrium height (or porosity) of the protective layer.
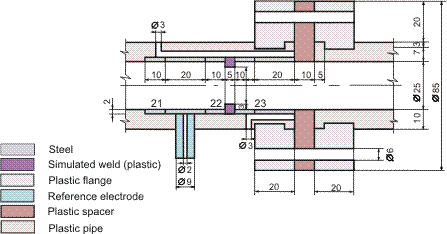
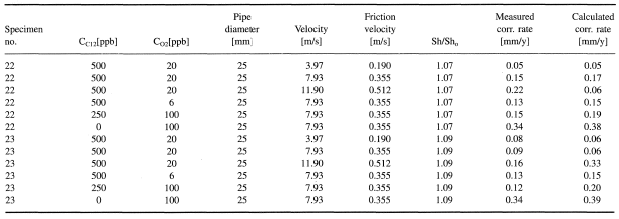
Figures 1-2 contain estimated magnitudes of the mass transfer expressed as Sherwood number (Sh) for geometries used in the corrosion tests [Soendvedt (1991)].
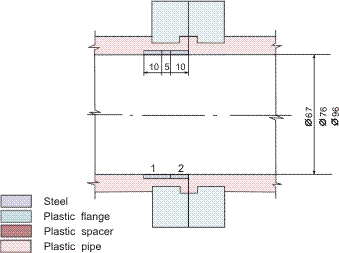
The main unknown in this model is the height of the protective layer. This must be determined by correlation with experiments. Derivation of apparent height characteristics for different geometries follows below.
The model presented above has been correlated with several experimental points.
If no chlorine is present, the calculated limiting corrosion rates fit with the recorded corrosion rates, i.e., no protective layer is found on the smooth sections.
To correlate with the experiments when chlorine is present, a protective layer must be introduced in the simulation of the experiments. The derived layer thickness is large and given as follows:

Sudden reduction in diameter:
The geometry considered is illustrated in Figures 3 and 4. The results are included in Table 3. The corrosion rates are significantly smaller than the limiting value.

Table 3. Cross-over (reduction in diameter)—test conditions and results from measurements and calculations

Analysis of the experiments reveals that the effective protective layer height varies with friction velocity as follows:

It has been observed that H is reduced as u* increases with an exponential of similar value to that given by Eq. (38) (i.e., u* to a power of 1.1).
The results are presented in Table 4.
The correlated protective layer height is described with the same type of formula as for sudden reduction in diameter:

H is zero when no chlorine is present. Again, the corrosion rates with Cl2 present are significantly smaller than the limiting values.
The geometry considered is sketched in Figure 5. The correlation for thickness of the protective layer is given by:
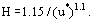
This leads to corrosion rates much smaller than magnitudes corresponding with no/inefficient protective layer (see Table 5).
The geometry in question is sketched in Figures 4 and 6.
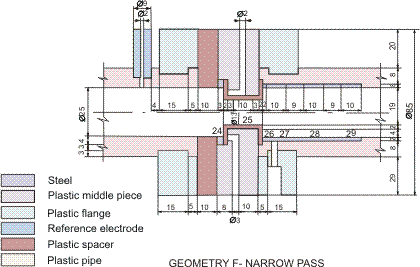
In the reversed flow zone, die increase of Sh/Sh0 is given as follows:

The high wall shear stress fluctuations in this region seem to give a thinner protective layer, H,
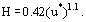
H is zero when no chlorine is present.
For the edge before the enlargement, the significant corrosion rates indicate a local high increase of mass transfer on the edge while the wall shear stress fluctuations should be small. To fit the data, Sh/Sb0 is given as follows:

Nomenclature
A Constant describing the pH dependence of surface reaction (-)
 Bulk concentration of species (mol/m3)
Bulk concentration of species (mol/m3)
cb, i Concentration of species i in the bulk (–)
cσ, i Concentration of species i at the diffusion layer boundary (-)
Cf Skin-friction coefficient
cs, i Concentration of species i at the surface (–)
CR Corrosion rate (mm/year)
D Pipe diameter (m)
Dmd, I Molecular diffusivity (m2/s)
E Electric field (V/m)
F Faraday constant (C/mol)
f Friction factor (–)
H Protective layer (μm)
HCO2 Henry constant
Ji Flux density of corrosive species i (mol/m3s)
Jconv, i Flux due to convective diffusion of species i (m/s)
ji Total flux of species i (m/s)
jmd, i Flux due to molecular diffusion of species i (m/s)
jmigr, i Flux due to migration of species i (m/s)
jr Reaction flux at surface (m/s)
K Constant (–)
Kp Permeability of protective layer (–)
K2 Corrosion rate corresponding with unit cathodic diffusion limited current (A/m2)
PCO2 Partial CO2 pressure (N/m2)
pHs pH at surface (–)
R Universal gas constant (J/K mol)
Re Reynolds number (–)
Sh Sherwood number (–)
Sh0 Sherwood number for smooth pipe (–)
T Bulk fluid temperature (°C)
U∞ Pipe velocity (m/s)
u Velocity field (m/s)
u Velocity component in x-direction (m/s)
u* Friction velocity (m/s)
v Velocity component in y-direction (m/s)
w Velocity component in z-direction (m/s)
x x coordinate (m)
y y coordinate (m)
z z coordinate (m)
zi Valency of ion i (–)
Greek Symbols
βm Mass transfer coefficient (m/s)
εD, i Eddy diffusivity (m2/s)
δdl Diffusion layer thickness (m)
δvl Viscous sub layer thickness (m)
γ Constant
λi Limiting ionic conductance of species i in water at 298 K (m2C2/J mol s)
φ Electric potential (V)
ρ Liquid density (kg/m3)
ν Kinematic viscosity (m2/s)
τW Wall shear on the layer (N/m2).
REFERENCES
Andersen, T. R. and Soendvedt, T. (1991) The influence of chlorine, oxygen and flow on corrosion in seawater injection systems, UK Corrosion Manchester Conference 22-24 Oct.
DeWaard, C. and Milliams, D. E. (1975) "Carbonic Acid Corrosion of Steel". Corrosion, 31, 177-181.
Dugstad, A. and Videm, K. (1990) Kjeller Sweet Corrosion-II Final Report IFE/KR/F-90/008.
Edwards, T. J., Maurer, G., Newman, J., and Prausnitz, J. M. (1978) Vapor-liquid Equilibrium in Multicomponent Aqueous Solutions of Volatile Weak Electrolytes, AlChE Journal, 24, 996-976.
Eriksrud, E. (1980) Internal Corrosion of Offshore Pipelines, Veritas Report No. 80-0517.
Fischer, W. and Siedlarek, W. (1978) "Werkstoffe und Korrosion" 28, 822.
Gundersen, R., et al. (1989) The effect of sodium hypochlorite on the electrochemical properties of stainless steel and on the bacterial activity in seawater. NACE Corrosion’ 89, Paper no 108, New Orleans, Louisiana, April 17-21.
Henriksen, N. et al. (1987) Efficient new processing system for water deoxidation. Off-shore Technology Conference, Paper no. OTC 5591, Houston, Texas, April 27-30.
Horner, R.A. et al. (1994) The Forties Water Injection System—A Review of Corrosion Control (Late Paper), UK Corrosion Wemberly Conference Centre 12-14 Nov. 1984.
Hudgins, C. M. (1971) The Oil and Gas Journal, February 15, 71.
Kharaka, Y. K., Gunter, W. D “ Aggarwal, P. K., Perins, E. H., and DeBraall, F. D. I. (1988) "SOLMINEQ88: A Computer Program for Geochemical Modelling of Water-Rock Interactions". U.S. Geological Survey, Water-Resources Investigations Report 88-4227, Menio Park-Ca.
Levich, V. G. (1962) Physicochemical Hydrodynamics. Prentice Hall, Inc.
Liu, G. and High, M. S. (1992) Final Report: Down hole Corrosion Modelling. A report to Norsk Hydro a.s. School of Chemical Engineering, Oklahoma State University.
Manner, R. and Heitz, E. (1978) Werkstoffe und Korrosion 29, 783.
Mitchell, R. W. (1978) Journal of Petroleum Technology, June, 887.
Newman, J. S. (1973) Electrochemical Systems. Prentice Hall, Engellewood Cliffs, N.J.
Nordgaard, A. and Soendvedt, T. (1994) Mechanistic CO2 corrosion modelling. Model formulation and comparisons with experiments up to 60 °C. Norsk Hydro Publication, Oil and Gas Division, Stabekk, Norway.
Patankar, S. V. (1980) Numerical Heat Transfer and Fluid Flow, Hemisphere publishing corporation, McGraw-Hill book company.
Sanchez-Caldera, L. E. (1984) The mechanism of corrosion-erosion in steam extraction lines of power plants, Massachusetts Institute of Technology.
Schmitt, G. and Rothmann, B. (1978) Werkstoffe und Korrosion 29, 98 and 237.
Schwenk, W. (1974) Werkstoffe und Korrosion, 25. 643.
Surman, P. L. and Castle, E. J. (1969) Corrosion Science, 9.
Soendvedt, T. (1991) Prediction of corrosion rate in deoxidized and chlorinated sea water. Development of model. Prod. Technology, U&P Division, Norsk Hydro.
References
- Andersen, T. R. and Soendvedt, T. (1991) The influence of chlorine, oxygen and flow on corrosion in seawater injection systems, UK Corrosion Manchester Conference 22-24 Oct.
- DeWaard, C. and Milliams, D. E. (1975) "Carbonic Acid Corrosion of Steel". Corrosion, 31, 177-181.
- Dugstad, A. and Videm, K. (1990) Kjeller Sweet Corrosion-II Final Report IFE/KR/F-90/008.
- Edwards, T. J., Maurer, G., Newman, J., and Prausnitz, J. M. (1978) Vapor-liquid Equilibrium in Multicomponent Aqueous Solutions of Volatile Weak Electrolytes, AlChE Journal, 24, 996-976. DOI: 10.1002/aic.690240608
- Eriksrud, E. (1980) Internal Corrosion of Offshore Pipelines, Veritas Report No. 80-0517.
- Fischer, W. and Siedlarek, W. (1978) "Werkstoffe und Korrosion" 28, 822.
- Gundersen, R., et al. (1989) The effect of sodium hypochlorite on the electrochemical properties of stainless steel and on the bacterial activity in seawater. NACE Corrosion’ 89, Paper no 108, New Orleans, Louisiana, April 17-21.
- Henriksen, N. et al. (1987) Efficient new processing system for water deoxidation. Off-shore Technology Conference, Paper no. OTC 5591, Houston, Texas, April 27-30.
- Horner, R.A. et al. (1994) The Forties Water Injection System—A Review of Corrosion Control (Late Paper), UK Corrosion Wemberly Conference Centre 12-14 Nov. 1984.
- Hudgins, C. M. (1971) The Oil and Gas Journal, February 15, 71.
- Kharaka, Y. K., Gunter, W. D “ Aggarwal, P. K., Perins, E. H., and DeBraall, F. D. I. (1988) "SOLMINEQ88: A Computer Program for Geochemical Modelling of Water-Rock Interactions". U.S. Geological Survey, Water-Resources Investigations Report 88-4227, Menio Park-Ca.
- Levich, V. G. (1962) Physicochemical Hydrodynamics. Prentice Hall, Inc.
- Liu, G. and High, M. S. (1992) Final Report: Down hole Corrosion Modelling. A report to Norsk Hydro a.s. School of Chemical Engineering, Oklahoma State University.
- Manner, R. and Heitz, E. (1978) Werkstoffe und Korrosion 29, 783.
- Mitchell, R. W. (1978) Journal of Petroleum Technology, June, 887.
- Newman, J. S. (1973) Electrochemical Systems. Prentice Hall, Engellewood Cliffs, N.J.
- Nordgaard, A. and Soendvedt, T. (1994) Mechanistic CO2 corrosion modelling. Model formulation and comparisons with experiments up to 60 °C. Norsk Hydro Publication, Oil and Gas Division, Stabekk, Norway.
- Patankar, S. V. (1980) Numerical Heat Transfer and Fluid Flow, Hemisphere publishing corporation, McGraw-Hill book company.
- Sanchez-Caldera, L. E. (1984) The mechanism of corrosion-erosion in steam extraction lines of power plants, Massachusetts Institute of Technology.
- Schmitt, G. and Rothmann, B. (1978) Werkstoffe und Korrosion 29, 98 and 237.
- Schwenk, W. (1974) Werkstoffe und Korrosion, 25. 643.
- Surman, P. L. and Castle, E. J. (1969) Corrosion Science, 9.
- Soendvedt, T. (1991) Prediction of corrosion rate in deoxidized and chlorinated sea water. Development of model. Prod. Technology, U&P Division, Norsk Hydro.